Синдром мальабсорбции – в каких случаях необходимо обратиться к гастроэнтерологу?
Синдром мальабсорбции — это хроническое заболевание, при котором плохо переваривается пища, а также происходит нарушение транспорта и всасывания витаминов и других полезных веществ в тонком кишечнике. Все это вызывает нарушение обмена веществ в организме.
Врожденный синдром мальабсорбции обнаруживается в 10% случаев; заболевание начинает проявляться сразу же после рождения или в первые 10 лет жизни. Также бывает приобретенный синдром мальабсорбции, который развивается в результате перенесенных заболеваний пищеварительной системы.
Специалисты разделяю синдром мальабсорбции по степени тяжести:
- Первая (легкая) степень.
Заболевание на этой стадии выражается в похудении до 10 кг, общей слабости, снижении работоспособности и некоторых признаках гиповитаминоза.
- Вторая (средней тяжести) степень
На этой стадии болезнь характеризуется потерей веса более, чем на 10 кг, выраженным гиповитаминозом, нарушением водно-электролитного обмена, анемией, снижением количества половых гормонов.
- Третья (тяжелая) степень
Характеризуется значительным дефицитом массы тела, тяжелой поливитаминной и электролитной недостаточностью, остеопорозом, выраженной анемией, отеками, судорогами, серьезными эндокринными нарушениями.
Синдром мальабсорбции: симптомы
Можно заподозрить заболевание, если плохо переваривается пища, беспокоят диарея, стеаторея, боли в животе, гиповитаминоз, потеря веса, астеновегетативный синдром, нарушения электролитного обмена, анемия.
При мальабсорбции происходят изменения со стороны кишечника, которые проявляются также вздутием живота и урчанием, иногда болями в животе. Зачастую боль наблюдается в верхней части живота, может отдавать в поясницу или имеет опоясывающий характер.
Кроме того, в большинстве случаев количество кала увеличено, испражнения имеют водянистую или кашеобразную консистенцию, обладают зловонным запахом. Если нарушается всасывание жирных кислот, кал обесцвечивается и имеет жирный блеск.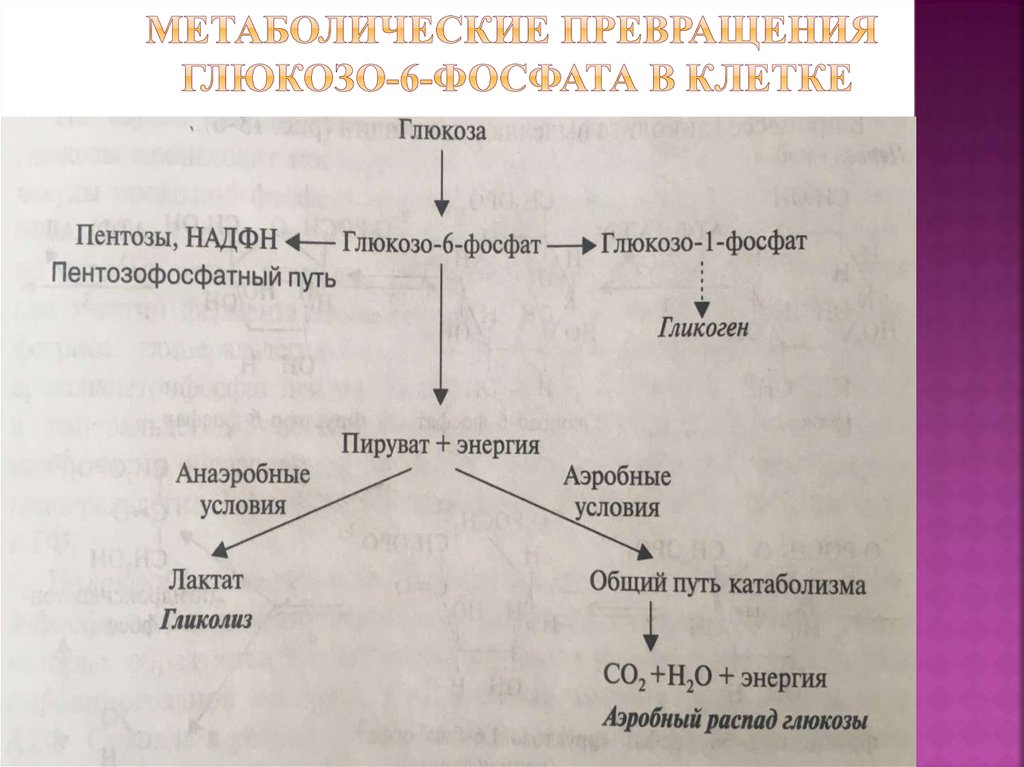
Также развивается астеновегетативный синдром — быстрая утомляемость, слабость, апатия. Это вызвано нарушением водно-электролитного обмена и дефицитом веществ, необходимых для нормального функционирования нервной системы.
Синдром мальабсорбции также характерен дерматитами, экземами, сухостью кожи, пигментацией, ломкостью ногтей, выпадением волос — они связаны с нехваткой витаминов и микроэлементов. По этим же причинам зачастую отмечаетсявоспаление языка.
При тяжелых нарушениях у больных наблюдаются выраженные отеки и скопление жидкости в брюшной полости.
Все пациенты с синдромом мальабсорбции склонны к снижению массы тела, которое с течением времени прогрессирует.
Дефицит витаминов ведет к различным расстройствам нервной системы, нарушениям зрения и анемии. Плохой обмен электролитов выражается в судорогах и мышечных болях.
Синдром мальабсорбции: диагностика
Диагностика заболевания основана на результатах общего и биохимического анализов крови, мочи, а также копрограммы, рентгенографии тонкого кишечника и УЗИ брюшной полости.
Осложнения мальабсорбции
Главные осложнения синдрома мальабсорбции грозят по причине нехватки питательных веществ: анемия, бесплодие, нервные расстройства, дистрофия. Также возможно развитие и других осложнений.
Лечение синдрома мальабсорбции
В легких случаях заболевание можно откорректировать при помощи диеты. А в остальных случаях лечение напрямую зависит от течения заболевания, выраженности нарушений всасывания и поступления веществ в кровь.
Если основная причина, вызвавшая развитие этого синдрома, устранена, коррекция последствий длительной дистрофии может занять много времени. Прогрессирование синдрома мальабсорбции грозит развитием опасных состояний и может привести даже к смерти.
Вот почему крайне важно верно установить диагноз и своевременно начать лечение мальабсорбции.
Терапия заболевания направлена на устранение причин мальабсорбции, а также витаминной, белковой, микроэлементной и электролитной недостаточности, дисбактериоза.
Обращайтесь в семейную клинику «Амеда» — наши лучшие специалисты смогут обеспечить высококвалифицированную диагностику и лечение заболевания.
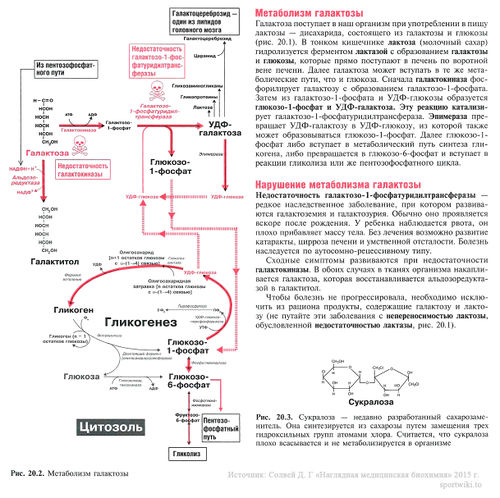
Глюкозо-галактозная мальабсорбция
Глюкозо-галактозная мальабсорбция – заболевание, которое связано с нарушением усвоения простых углеводов в кишечнике. Обусловлено оно дефектом транспортных систем щеточной каймы энтероцитов. Синдром глюкозо-галактозной мальабсорбции может быть врожденным (диагностируется при первых кормлениях новорожденных) и приобретенным (вызван другими болезнями ЖКТ).
Симптомы глюкозо-галактозной мальабсорбции
- диарея;
- астеновегетативный синдром;
- стеаторея.
Они проявляются после приема различных продуктов питания, в составе которых есть крахмал, лактоза, сахароза, мальтоза или моносахариды (кроме фруктозы). У многих больных развивается дегидратация, выраженная дисфункция кишечника и повышается температура тела.
Лечение глюкозо-галактозной мальабсорбции
Лечение глюкозо-галактозной мальабсорбции и заболеваний, возникших на ее фоне (сахарного диабета, дифицита лактозы и т. д.) – сложная задача, поскольку практически все продукты содержат дисахариды или моносахариды. При первичном типе такой болезни единственным углеводом, который способен всасываться в кишечнике, является фруктоза. Именно поэтому больному показано питание смесями, которые составлены на основе нескольких видов белка, и парентеральное введение глюкозы.
д.) – сложная задача, поскольку практически все продукты содержат дисахариды или моносахариды. При первичном типе такой болезни единственным углеводом, который способен всасываться в кишечнике, является фруктоза. Именно поэтому больному показано питание смесями, которые составлены на основе нескольких видов белка, и парентеральное введение глюкозы.
При вторичной глюкозо-галактозной мальабсорбции или дефиците лактазы пациент должен соблюдать строжайшую диету. Ему можно употреблять в пищу только продукты с высоким содержанием белка, пюре из фруктозо-содержащих овощей и растворы глюкозы. На различных этапах лечения возможен срыв адаптации ферментативных систем ЖКТ к любому углеводу или к повышению его объема. В таких случаях необходимо полностью исключить непереносимые продукты, но со временем можно снова попытаться с их помощью расширить рацион.
Статьи по теме:
Опоясывающая боль в области желудка и спины Практически все люди хотя бы раз в жизни испытали опоясывающая боль в животе и пояснице. |
Болят колени – что делать? Колени болят у многих. Коленный сустав самый крупный, поэтому ему приходится регулярно справляться с большими нагрузками. Иногда он выходит из строя. Зачастую колени болят очень сильно. Но есть кое-какие средства, которые могут убрать болезненные ощущения. |
|
Можно ли вылечить эпилепсию? В глазах многих людей эпилепсия – одно из самых страшных заболеваний, которые могут существовать в природе. Недуг в самом деле проявляется пугающими симптомами. Но он не такой уж и страшный. По крайней мере, если запастись терпением и поверить в себя, с ним можно справиться. | Болит колено при сгибании и разгибании Стараетесь не сгибать и не разгибать ноги в коленях из-за острой боли при данных движениях? Ищете причины этой проблемы, заболевания, которые ее провоцируют, и действенные способы лечения? Тогда вам нужно обязательно прочитать нашу сегодняшнюю статью. |
 В большинстве случаев такие ощущения вызваны растяжение мышц или физическим переутомлением. Но иногда они свидетельствуют о заболевании внутренних органов. Подробнее об этом – в нашей статье.
В большинстве случаев такие ощущения вызваны растяжение мышц или физическим переутомлением. Но иногда они свидетельствуют о заболевании внутренних органов. Подробнее об этом – в нашей статье.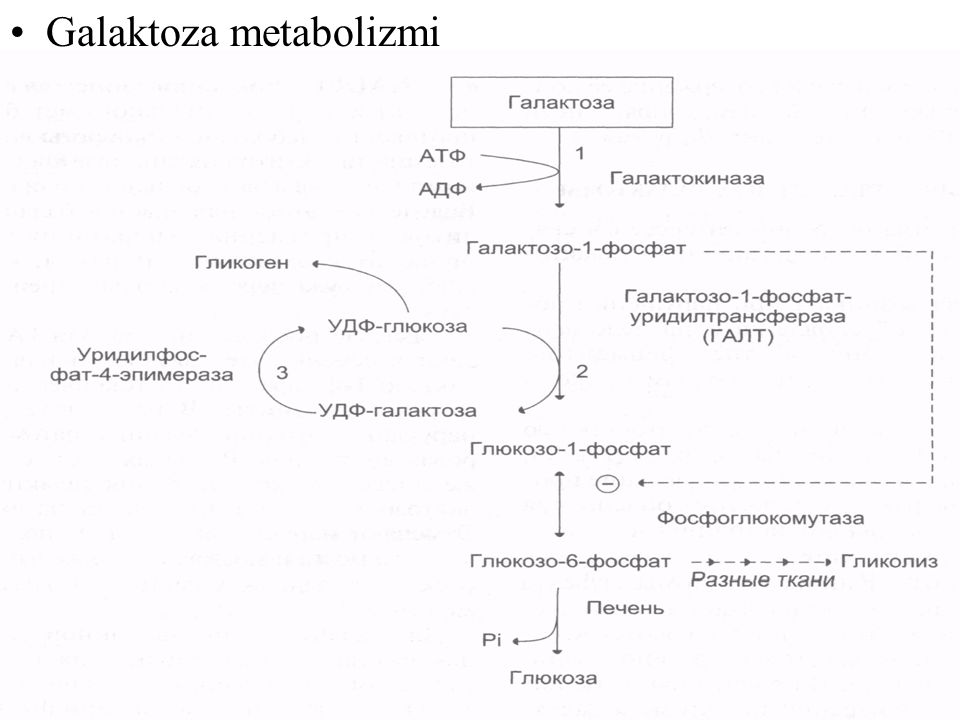
Врожденная глюкозо-галактозная мальабсорбция: случай с новой мутацией SLC5A1 у ребенка из Саудовской Аравии
- Статья
- Авторы и т.д.
- Метрики
- Цифры и т.д.
Луджен О. Аламуди, Албараа Т. Альфараиди, Самия С. Альтагафи, Маджид С. Аль-Такафи , Мохаммед Хасоса
Опубликовано: 02 октября 2021 г. (см. историю)
DOI: 10.7759/куреус.18440
Цитируйте эту статью как: Аламуди Л.О., Альфараиди А.Т., Альтагафи С.С. и др. (02 октября 2021 г.) Врожденная глюкозо-галактозная мальабсорбция: случай с новой мутацией SLC5A1 у младенца из Саудовской Аравии. Куреус 13(10): e18440. doi: 10.7759/cureus.18440
Abstract
Хотя в педиатрии было зарегистрировано всего несколько сотен случаев, врожденная глюкозо-галактозная мальабсорбция (GGM) является чрезвычайно редким аутосомно-рецессивным нарушением обмена веществ, характеризующимся трудноизлечимой диареей и тяжелой дегидратацией, которая может быть пожизненной. угроза, если не лечить должным образом. Из-за редкости заболевания большинству клиницистов сложно рассматривать ГГМ в качестве первоначального диагноза. Мы сообщаем о клиническом и диагностическом течении семимесячного младенца из Саудовской Аравии, у которого наблюдались тяжелые повторяющиеся эпизоды водянистой диареи и задержка развития в раннем младенчестве, несмотря на стандартное лечение. Молекулярное тестирование показало, что у нашего пациента был сложный гетерозиготный вариант в SLC5A1 . Было доказано, что формулы на основе фруктозы эффективны при лечении GGM. Этот случай подчеркивает важность ранней диагностики и своевременного лечения для предотвращения серьезных осложнений невыявленного ГГМ.
угроза, если не лечить должным образом. Из-за редкости заболевания большинству клиницистов сложно рассматривать ГГМ в качестве первоначального диагноза. Мы сообщаем о клиническом и диагностическом течении семимесячного младенца из Саудовской Аравии, у которого наблюдались тяжелые повторяющиеся эпизоды водянистой диареи и задержка развития в раннем младенчестве, несмотря на стандартное лечение. Молекулярное тестирование показало, что у нашего пациента был сложный гетерозиготный вариант в SLC5A1 . Было доказано, что формулы на основе фруктозы эффективны при лечении GGM. Этот случай подчеркивает важность ранней диагностики и своевременного лечения для предотвращения серьезных осложнений невыявленного ГГМ.
Введение
Глюкозо-галактозная мальабсорбция (GGM) (OMIM: 606824) — чрезвычайно редкое аутосомно-рецессивное нарушение обмена веществ, которое характеризуется дефектом транспорта глюкозы и галактозы через границу кишечника [1]. Люди с GGM несут две копии дефектного гена SLC5A1, расположенного в хромосоме 22q13. 1. У здоровых младенцев кишечный натрий-зависимый котранспортер глюкозы (SGLT1) направляет глюкозу и воду против градиентов их концентрации в энтероциты посредством Na+ и электрических градиентов [2]. Мутации в гене SLC5A1 приводят к дефекту SGLT1 через щеточную кайму кишечника, что приводит к накоплению неабсорбированного натрия, глюкозы и галактозы внутри просвета кишечника [3]. GGM характеризуется неонатальным началом тяжелой водянистой диареи, которая без надлежащего лечения приводит к летальному исходу в течение нескольких недель [2]. Несмотря на то, что от ГГМ нет лекарства, с ним можно справиться, исключив из рациона лактозу, глюкозу и сахарозу. Младенцы, у которых пренатально диагностирован ГГМ, могут лучше питаться смесью на основе фруктозы, а впоследствии будут продолжать придерживаться твердой диеты на основе фруктозы [1]. Эпидемиологические данные по ГГМ отсутствуют, с 2001 по 2019 год зарегистрировано всего 107 случаев.во всем мире [3], и клиницистам сложно рассматривать ГГМ в качестве первоначального диагноза.
1. У здоровых младенцев кишечный натрий-зависимый котранспортер глюкозы (SGLT1) направляет глюкозу и воду против градиентов их концентрации в энтероциты посредством Na+ и электрических градиентов [2]. Мутации в гене SLC5A1 приводят к дефекту SGLT1 через щеточную кайму кишечника, что приводит к накоплению неабсорбированного натрия, глюкозы и галактозы внутри просвета кишечника [3]. GGM характеризуется неонатальным началом тяжелой водянистой диареи, которая без надлежащего лечения приводит к летальному исходу в течение нескольких недель [2]. Несмотря на то, что от ГГМ нет лекарства, с ним можно справиться, исключив из рациона лактозу, глюкозу и сахарозу. Младенцы, у которых пренатально диагностирован ГГМ, могут лучше питаться смесью на основе фруктозы, а впоследствии будут продолжать придерживаться твердой диеты на основе фруктозы [1]. Эпидемиологические данные по ГГМ отсутствуют, с 2001 по 2019 год зарегистрировано всего 107 случаев.во всем мире [3], и клиницистам сложно рассматривать ГГМ в качестве первоначального диагноза.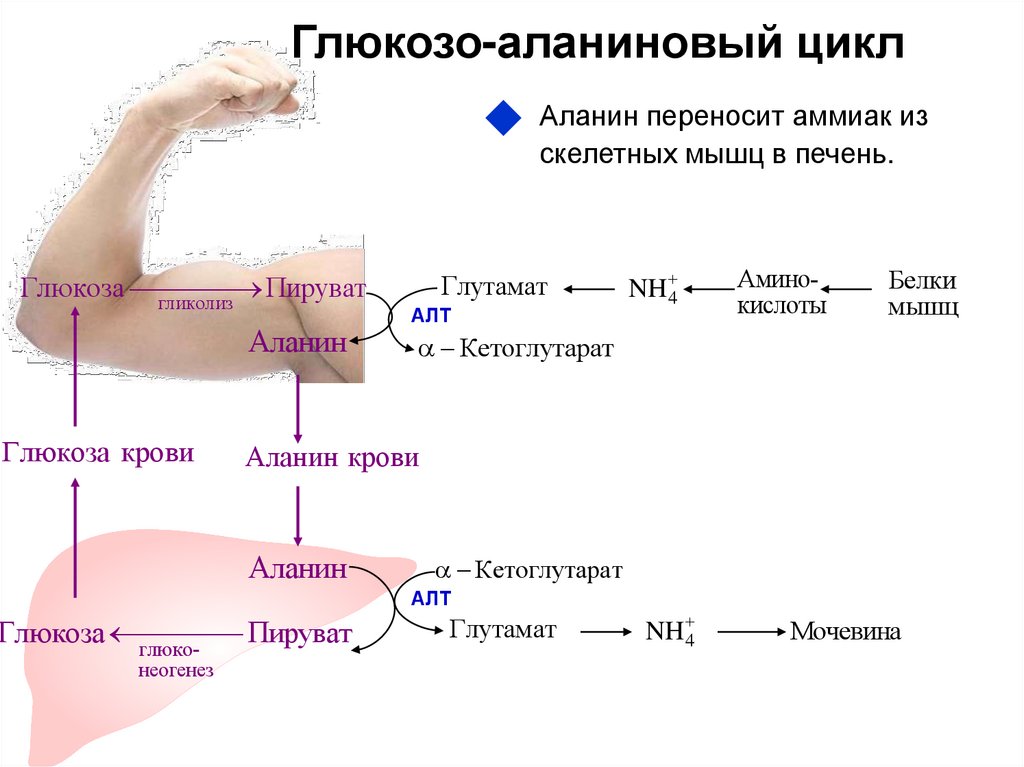 Здесь мы представляем клиническое и диагностическое течение саудовского младенца с GGM, у которого обнаружена гетерозиготная мутация в гене SLC5A1.
Здесь мы представляем клиническое и диагностическое течение саудовского младенца с GGM, у которого обнаружена гетерозиготная мутация в гене SLC5A1.
Презентация клинического случая
Здесь мы представляем случай семимесячного мальчика, у которого произошли спонтанные вагинальные роды в срок; беременность, роды и роды протекали без осложнений. Он родился от некровных родителей и имеет двух старших здоровых братьев и сестер без значимой семейной истории. Младенец несколько раз обращался в отделение неотложной помощи по поводу эпизодов тяжелой диареи, которые начались в возрасте четырех месяцев. Водянистый стул около 10-15 раз в день без улучшения, не связанного с острым заболеванием. Первоначально пациент находился на исключительно искусственном вскармливании, но его симптомы значительно усилились после введения твердой пищи в возрасте шести месяцев. В анамнезе не было рвоты, плохого питания или рецидивирующих инфекций. Его развитие и прививки соответствовали его возрасту.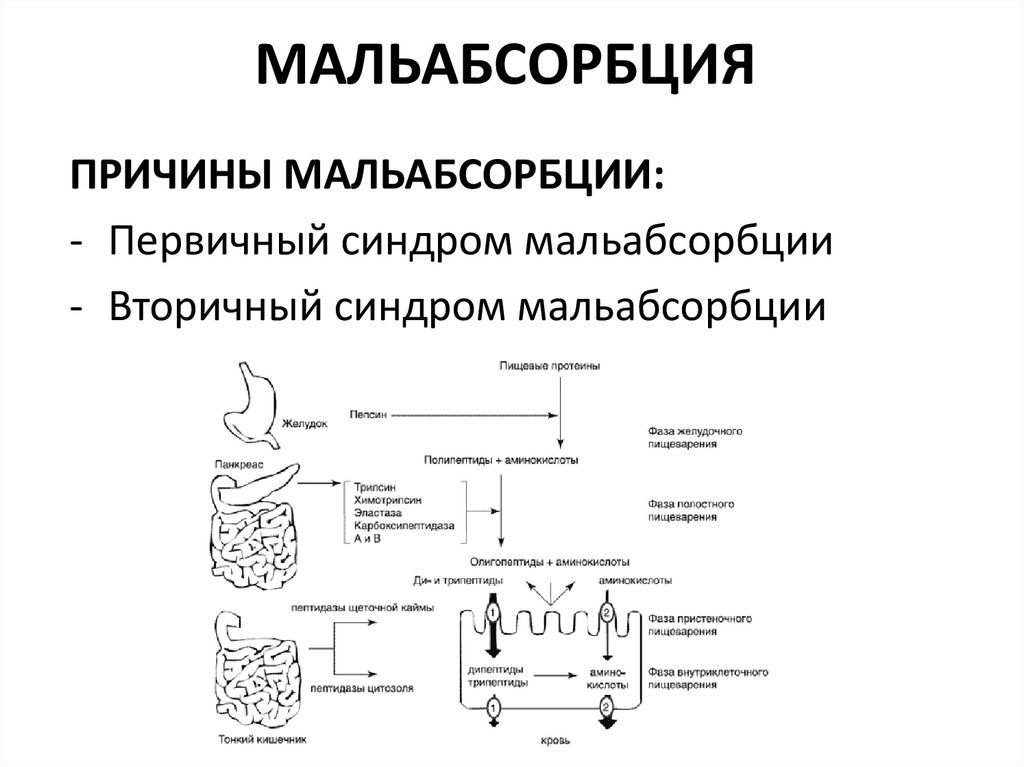
При медицинском осмотре у него был недостаточный вес, он выглядел худым и бледным. Его вес был ниже третьего процентиля со значительно выступающим животом, но не было отмечено гепатомегалии или спленомегалии. Физикальное обследование других систем ничем не примечательно. Он прошел обширное диагностическое обследование, включая базовые рутинные лабораторные анализы, тест на хлориды пота, антитканевую трансглютаминазу IgA, тест на пищевую аллергию и коровье молоко, а также эндоскопию верхних отделов желудочно-кишечного тракта, которые прошли нормально без отклонений, как показано в Таблице 9.0049 1 . Нейтропению у нашего ребенка можно объяснить дефицитом фолиевой кислоты и витамина В12 (уровни этих минералов в сыворотке не показаны в таблице). Различные диетические ограничения, включая безглютеновую диету, были опробованы без улучшения.
| Название теста | Результат | Блок | Диапазон задания |
| Средняя корпускулярная концентрация Hgb | 33,4 | г/дл | 31,9-35,2 |
| Число тромбоцитов | 223 | 10 9 /л | 140-450 |
| Средний объем тромбоцитов | 10,7 | фл | 8,6-12,3 |
| Эозинофилы # | 0,01 | 10 9 /л | 0,1-1,1 |
| Эритроциты с ядром % | 0,0 | % | 0-0 |
| Подсчет эритроцитов | 5,41 | 10 12 /л | 3,7-5,3 |
| Ширина распределения эритроцитов | 13,9 | % | 11,5-15,3 |
| Лимфоциты # | 4,78 | 10 9 /л | 2,5-13,5 |
| Базофилы # | 0,01 | 10 9 /л | 0-0,1 |
| Гемоглобин | 13,7 | г/дл | 10,5-13,5 |
| Гематокрит | 41,0 | % | 33-39 |
| Средний корпускулярный объем | 75,8 | фл | 70-86 |
| Среднее корпускулярное значение Hgb | 25,3 | стр | 23-31 |
| Подсчет лейкоцитов | 5,82 | 10 9 /л | 5,5-17 |
| Нейтрофилы # | 0,66 | 10 9 /л | 1,5-10 |
| Моноциты # | 0,36 | 10 9 /л | 0,2-1,1 |
Стол 1: Полный анализ крови с дифференциалом
Hgb, гемоглобин; Эритроциты, эритроциты
Геномная ДНК была выделена из периферической крови и отправлена на полноэкзомное секвенирование (WES).
Обсуждение
Ген SLC5A1 кодирует SGLT1, расположенный на щеточной кайме эпителия кишечника [4]. SGLT1 отвечает за перемещение глюкозы, аминокислот, витаминов и некоторых ионов через эпителий кишечника [5]. GGM возникает вторично из-за дефекта этого кишечного SGLT1; однако на всасывание фруктозы и ксилозы это не влияет. Накопление неабсорбированного натрия, глюкозы и галактозы в просвете кишечника приводит к осмотической диарее и тяжелой дегидратации. Это может привести к летальному исходу у новорожденных с ГГМ, если своевременно не будет оказано соответствующее лечение. Непереносимость углеводов по генетическим причинам может иметь раннее или позднее начало. Непереносимость углеводов с ранним началом включает GGM, врожденный дефицит сахаразы-изомальтазы и врожденный дефицит лактазы. Непереносимость углеводов с поздним началом включает непереносимость лактозы взрослого типа [6].
Это может привести к летальному исходу у новорожденных с ГГМ, если своевременно не будет оказано соответствующее лечение. Непереносимость углеводов по генетическим причинам может иметь раннее или позднее начало. Непереносимость углеводов с ранним началом включает GGM, врожденный дефицит сахаразы-изомальтазы и врожденный дефицит лактазы. Непереносимость углеводов с поздним началом включает непереносимость лактозы взрослого типа [6].
ГГМ следует заподозрить, если присутствуют следующие признаки: (1) водянистая диарея с началом вскоре после рождения; (2) признаки мальабсорбции углеводов с положительными восстанавливающими веществами в кале; (3) отсутствие улучшения при использовании безлактозных и аминокислотных смесей; (4) улучшение диареи только при элиминации глюкозы и галактозы; (5) исключение инфекций [2,6]. Испытания различных молочных смесей, в том числе безлактозно-галактозных смесей, объясняют позднее обращение нашего пациента. Кроме того, известно, что популяция, из которой произошел ребенок, имеет GGM в этом возрасте [7].
Более 40 вариантов SLC5A1 связаны с GGM [8]. WES нашего пациента выявила патогенный гетерозиготный вариант c.765C>G p.(Cys255Trp) в гене SLC5A1. Ранее в литературе сообщалось, что этот вариант вызывает GGM [4]. Другой гетерозиготный вариант был идентифицирован c.899G>A p. (Arg300His) в SLC5A1 (OMIM: 182380), который, как сообщается, вероятно является патогенным; однако ранее он не был описан в литературе.
Родство между родителями играет важную роль в наследовании вызывающей болезнь мутации, поскольку ГГМ является аутосомно-рецессивным заболеванием. Lee WS сообщил о первом случае GGM в Малайзии в 1997 [9]. Отсутствуют данные о заболеваемости или распространенности этого заболевания. Недавний обзор Wang et al. сообщили, что из 107 рассмотренных случаев 55 случаев были из Саудовской Аравии и Турции, что составляет 78,2 % от общего числа опубликованных случаев [3]. Врачи должны проявлять бдительность в тех странах, когда у младенца наблюдается неизлечимая обильная водянистая диарея и обезвоживание, сопровождающееся задержкой развития, которые сохраняются независимо от стандартной терапии.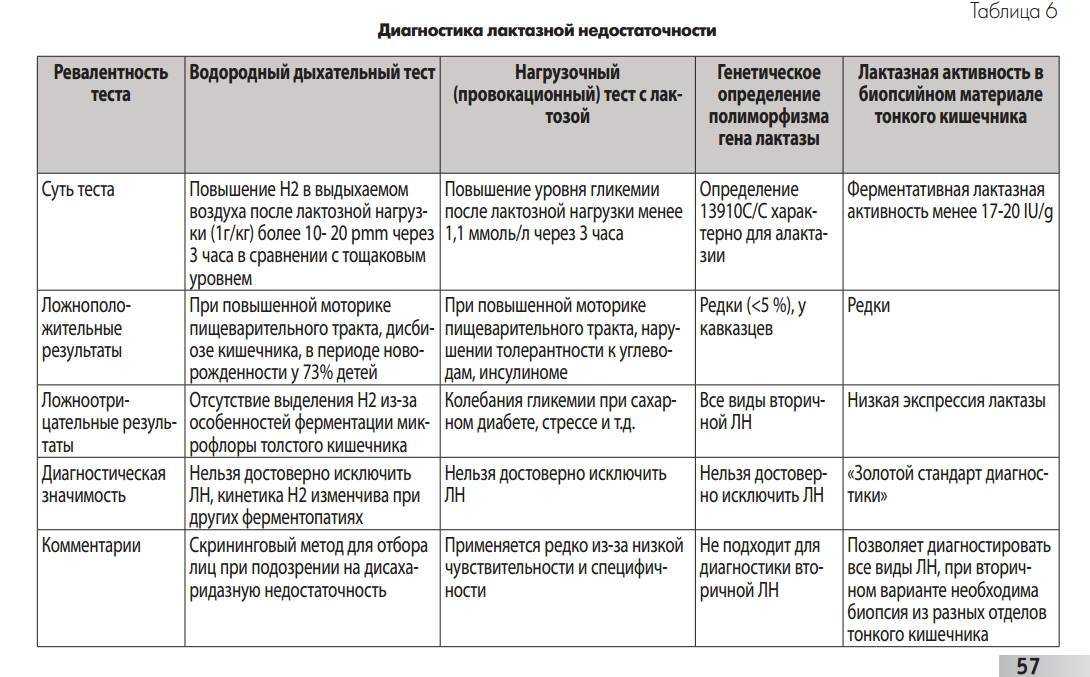 Хотя у младенцев с GGM возникает водянистая диарея после начала грудного вскармливания в течение нескольких дней после рождения, могут присутствовать такие симптомы, как вздутие живота и колики [10]. Кроме того, у пациентов с GGM может наблюдаться перемежающаяся глюкозурия, что было отмечено у пакистанского младенца [11].
Хотя у младенцев с GGM возникает водянистая диарея после начала грудного вскармливания в течение нескольких дней после рождения, могут присутствовать такие симптомы, как вздутие живота и колики [10]. Кроме того, у пациентов с GGM может наблюдаться перемежающаяся глюкозурия, что было отмечено у пакистанского младенца [11].
Если не лечить, у младенцев может развиться нефролитиаз и нефрокальциноз из-за хронического обезвоживания, что было зарегистрировано в серии случаев пяти арабских детей с GGM. В результате гипернатриемии и обезвоживания у одного из детей развилась гангрена обеих ног, что потребовало двусторонней ампутации [10]. Камни в почках также наблюдались и описаны Meeuwisse в 1969 году; однако их причина до сих пор неизвестна [12].
Что касается диагностики, то измерение концентрации водорода в выдыхаемом воздухе после употребления глюкозы может быть диагностическим; однако у младенцев он ненадежен. Полная ремиссия диарейных симптомов после полного исключения глюкозы и галактозы из рациона может быть использована для предварительного диагноза ГГМ, что было адекватно продемонстрировано в случае малазийского младенца [13]. Водородный дыхательный тест с глюкозой или галактозой или пероральный тест на толерантность к глюкозе/галактозе был заменен современным генетическим тестированием. Биопсия тонкой кишки с нормальной гистопатологией может исключить другие причины мальабсорбции; однако в этом нет необходимости, если диагноз можно подтвердить с помощью менее инвазивных тестов. Идентификация мутаций в гене SLC5A1 с помощью WES подтверждает диагноз [14].
Водородный дыхательный тест с глюкозой или галактозой или пероральный тест на толерантность к глюкозе/галактозе был заменен современным генетическим тестированием. Биопсия тонкой кишки с нормальной гистопатологией может исключить другие причины мальабсорбции; однако в этом нет необходимости, если диагноз можно подтвердить с помощью менее инвазивных тестов. Идентификация мутаций в гене SLC5A1 с помощью WES подтверждает диагноз [14].
Диагностика ГГМ затруднена из-за редкости заболевания. Высокая клиническая подозрительность имеет важное значение, и после подтверждения диагноза GGM необходимо немедленно начать лечение. Формула на основе фруктозы показала себя как эффективное лечение с резкой реакцией у младенцев, приводя к прекращению эпизодов диареи и способствуя нормальному неврологическому развитию и росту. Обучение родителей в отношении управления питанием GGM является важным компонентом диетотерапии младенцев [15]. Генетическое консультирование в отношении будущих детей может быть предложено родителям, в семейном анамнезе которых есть новорожденные, пораженные ГГМ, особенно при наличии кровного родства. Прогноз у младенцев с ГГМ лучше, если диагноз был установлен в раннем неонатальном периоде, поскольку ожидается, что с возрастом непереносимость глюкозы и галактозы улучшится [15]. Долгосрочное управление питанием остается сложным процессом, требующим соблюдения специального диетического плана и мониторинга долгосрочных последствий соблюдения диеты с высоким содержанием белков и жиров.
Прогноз у младенцев с ГГМ лучше, если диагноз был установлен в раннем неонатальном периоде, поскольку ожидается, что с возрастом непереносимость глюкозы и галактозы улучшится [15]. Долгосрочное управление питанием остается сложным процессом, требующим соблюдения специального диетического плана и мониторинга долгосрочных последствий соблюдения диеты с высоким содержанием белков и жиров.
Выводы
Ребенок был бессимптомным на смеси с добавлением фруктозы. За ним внимательно следили специалисты-гастроэнтерологи и диетолог, которые постоянно поддерживали и обучали его родителей. Раннее начало хронической диареи у пациентов с отрицательным результатом обследования на наиболее распространенные причины хронической диареи должно насторожить педиатра, чтобы он задумался о редких врожденных причинах хронической диареи. Настоятельно рекомендуется генетическое тестирование, которое помогает выявить таких пациентов раньше, тем самым предотвращая серьезные осложнения и улучшая клинические результаты.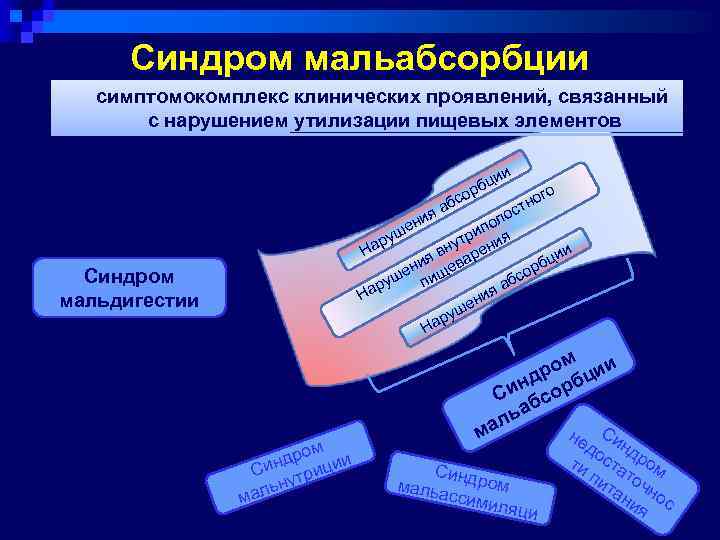
Ссылки
- Национальный центр биотехнологической информации (США): гены и болезни. Национальный центр биотехнологической информации (США), Бетесда, Мэриленд; 1998.
- Райт EM: I. Мальабсорбция глюкозы-галактозы. Am J Physiol. 1998, 275:G879-82. 10.1152/ajpgi.1998.275.5.G879
- Wang W, Wang L, Ma M: Обзор литературы о врожденной глюкозо-галактозной мальабсорбции с 2001 по 2019 год. J Paediatr Child Health. 2020, 56:1779-84. 10.1111/jpc.14702
- Райт Э.М., Терк Э., Мартин М.Г.: Молекулярная основа глюкозо-галактозной мальабсорбции. Клеточная биохимия Биофиз. 2002, 36:115-21. 10.1385/CBB:36:2-3:115
- Sabino-Silva R, Mori RC, David-Silva A, Okamoto MM, Freitas HS, Machado UF: Котранспортеры Na(+)/глюкозы: от генов к терапии. Braz J Med Biol Res. 2010, 43:1019-26. 10.1590/s0100-879×2010007500115
- Berni Canani R, Pezzella V, Amoroso A, Cozzolino T, Di Scala C, Passariello A: Диагностика и лечение непереносимости углеводов у детей.
 Питательные вещества. 2016, 8:157. 10.3390/nu8030157
Питательные вещества. 2016, 8:157. 10.3390/nu8030157 - Saadah OI, Alghamdi SA, Sindi HH, Alhunaitti H, Bin-Taleb YY, Alhussaini BH: Врожденная глюкозо-галактозная мальабсорбция: описательное исследование клинических характеристик и результатов из Западной Саудовской Аравии. Араб Дж. Гастроэнтерол. 2014, 15:21-3. 10.1016/j.ajg.2014.01.004
- Xin B, Wang H: Множественные вариации последовательности в гене SLC5A1 связаны с нарушением всасывания глюкозы-галактозы у большой когорты амишей старого порядка. Клин Жене. 2011, 79:86-91. 10.1111/j.1399-0004.2010.01440.x
- Lee WS, Goh AYT: Врожденная глюкозо-галактозная мальабсорбция – история болезни. ЮММЕК. 1997, 2:43-5.
- Ассири А., Саид А., Алнимри А., Ахмад С., Саид Э., Джамиль С. Пять арабских детей с глюкозо-галактозной мальабсорбцией. Педиатр Int Child Health. 2013, 33:108-10. 10.1179/2046
3Y.0000000055 - Rafeey M, Golzar A: Синдром глюкозо-галактозной мальабсорбции, проявляющийся врожденной диареей.
Пак J Med Sci. 2007, 23:959-61.- Meeuwisse GW, Melin K: глюкозо-галактозная мальабсорбция. Клиническое исследование 6 случаев. Acta Pediatr Scand. 1969, 1+.
- Lee WS, Tay CG, Nazrul N, Paed M, Chai PF: случай неонатальной диареи, вызванной врожденной глюкозо-галактозной мальабсорбцией. Med J Малайзия. 2009, 64:83-5.
- Андерсон С., Кониарис С., Синь Б., Брукс С.С.: Врожденная глюкозо-галактозная мальабсорбция: клинический случай. J Педиатр Здравоохранение. 2017, 31:506-10. 10.1016/j.pedhc.2017.01.005
- Абад-Синден А., Боровиц С., Мейерс Р., Сутфен Дж. Кормление при врожденной глюкозо-галактозной мальабсорбции: тематическое исследование. J Am Diet Assoc. 1997, 97:1417-21. 10.1016/s0002-8223(97)00342-8
Врожденная глюкозо-галактозная мальабсорбция: случай с новой мутацией SLC5A1 у ребенка из Саудовской Аравии
Информация об авторе
Лухен О. Аламуди
Медицинский колледж, Университет медицинских наук имени короля Сауда бин Абдулазиза, Международный медицинский исследовательский центр имени короля Абдуллы, Медицинский городок имени короля Абдулазиза, Министерство национальной гвардии – по вопросам здравоохранения, Джидда, SAU
Альбараа Т.
АльфараидиМедицинский колледж, Университет медицинских наук имени короля Сауда бин Абдулазиза, Международный медицинский исследовательский центр имени короля Абдуллы, Медицинский городок имени короля Абдулазиза, Министерство национальной гвардии – по вопросам здравоохранения, Джидда, SAU
Самия С. Альтагафи
Педиатрия, Военный госпиталь Аль-Хада, Таиф, SAU
Маджид С. Аль-Такафи Соответствующий автор
Профилактика и контроль инфекций, Министерство национальной гвардии – по вопросам здравоохранения, Джидда, SAU
Медицинский колледж, Университет медицинских наук имени короля Сауда бин Абдулазиза, Международный медицинский исследовательский центр имени короля Абдаллы, Медицинский городок имени короля Абдулазиза, Министерство национальной гвардии – по вопросам здравоохранения, Джидда, SAU
Мохаммед Хасоса
Детская гастроэнтерология, Министерство национальной гвардии – по вопросам здравоохранения, Джидда, SAU
Медицинский колледж, Университет медицинских наук имени короля Сауда бин Абдулазиза, Международный медицинский исследовательский центр имени короля Абдуллы, Медицинский город короля Абдулазиза, Министерство национальной гвардии – по вопросам здравоохранения, Джидда, SAU
Заявление об этике и раскрытие информации о конфликте интересов
Люди: Согласие было получено или от него отказались все участники этого исследования.
Конфликт интересов: В соответствии с единой формой раскрытия информации ICMJE все авторы заявляют следующее: Информация об оплате/услугах: Все авторы заявили, что никакая финансовая поддержка представленной работы не была получена ни от одной организации. Финансовые отношения: Все авторы заявили, что у них нет финансовых отношений в настоящее время или в течение предыдущих трех лет с какими-либо организациями, которые могут быть заинтересованы в представленной работе. Другие отношения: Все авторы заявили об отсутствии других отношений или действий, которые могли бы повлиять на представленную работу.Благодарности
Мы хотели бы поблагодарить пациента и его семью за их желание и сотрудничество принять участие в этом исследовании.
Информация о артикуле
ДОИ
10.7759/куреус.18440
Цитируйте эту статью как:
Аламуди Л.О., Альфараиди А.Т., Альтагафи С.
С. и др. (02 октября 2021 г.) Врожденная глюкозо-галактозная мальабсорбция: случай с новой мутацией SLC5A1 у младенца из Саудовской Аравии. Куреус 13(10): e18440. Дои: 10.7759/cureus.18440История публикаций
Начало экспертной оценки: 24 июля 2021 г.
Экспертная проверка завершена: 28 сентября 2021 г.
Опубликовано: 02 октября 2021 г.Авторское право
© Copyright 2021
Alamoudi et al. Это статья с открытым доступом, распространяемая в соответствии с лицензией Creative Commons Attribution License CC-BY 4.0., которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания оригинального автора и источника.Лицензия
Это статья с открытым доступом, распространяемая в соответствии с условиями лицензии Creative Commons Attribution License, которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии указания автора и источника.
Врожденная глюкозо-галактозная мальабсорбция: случай с новой мутацией SLC5A1 у ребенка из Саудовской Аравии
Цифры и т. д.
Результат Блок Диапазон задания Средняя корпускулярная концентрация Hgb 33,4 г/дл 31,9-35,2 Число тромбоцитов 223 10 9 /л 140-450 Средний объем тромбоцитов 10,7 фл 8,6-12,3 Эозинофилы # 0,01 10 9 /л 0,1-1,1 Эритроциты с ядром % 0,0 % 0-0 Подсчет эритроцитов 5,41 10 12 /л 3,7-5,3 Ширина распределения эритроцитов 13,9 % 11,5-15,3 Лимфоциты # 4,78 10 9 /л 2,5-13,5 Базофилы # 0,01 10 9 /л 0-0,1 Гемоглобин 13,7 г/дл 10,5-13,5 Гематокрит 41,0 % 33-39 Средний корпускулярный объем 75,8 фл 70-86 Среднее корпускулярное значение Hgb 25,3 стр 23-31 Количество лейкоцитов 5,82 10 9 /л 5,5-17 Нейтрофилы # 0,66 10 9 /л 1,5-10 Моноциты # 0,36 10 9 /л 0,2-1,1 Стол 1: Полный анализ крови с дифференциалом
Hgb, гемоглобин; Эритроциты, эритроциты
Посмотреть крупнее
Название теста Результат Блок Диапазон задания Средняя корпускулярная концентрация Hgb 33,4 г/дл 31,9-35,2 Число тромбоцитов 223 10 9 /л 140-450 Средний объем тромбоцитов 10,7 фл 8,6-12,3 Эозинофилы # 0,01 10 9 /л 0,1-1,1 Эритроциты с ядром % 0,0 % 0-0 Подсчет эритроцитов 5,41 10 12 /л 3,7-5,3 Ширина распределения эритроцитов 13,9 % 11,5-15,3 Лимфоциты # 4,78 10 9 /л 2,5-13,5 Базофилы # 0,01 10 9 /л 0-0,1 Гемоглобин 13,7 г/дл 10,5-13,5 Гематокрит 41,0 % 33-39 Средний корпускулярный объем 75,8 фл 70-86 Среднее корпускулярное значение Hgb 25,3 стр. 23-31 Подсчет лейкоцитов 5,82 10 9 /л 5,5-17 Нейтрофилы # 0,66 10 9 /л 1,5-10 Моноциты # 0,36 10 9 /л 0,2-1,1 —
ОЦЕНКА 1 ЧИТАТЕЛЯ
ОЦЕНКА СООБЩЕНИЯ
Scholarly Impact Quotient™ (SIQ™) — это наш уникальный процесс оценки рецензирования после публикации. Узнайте больше здесь.
Что такое SIQ™?
Scholarly Impact Quotient™ (SIQ™) — это наш уникальный процесс оценки рецензирования после публикации. SIQ™ оценивает важность и качество статей, используя коллективный разум сообщества Cureus в целом. Всем зарегистрированным пользователям предлагается внести свой вклад в SIQ™ любой опубликованной статьи. (Авторы не могут оценивать свои собственные статьи.)
Высокие рейтинги должны быть зарезервированы за работу, которая является действительно новаторской в соответствующей области.
Все, что выше 5, следует считать выше среднего. Хотя все зарегистрированные пользователи Cureus могут оценивать любую опубликованную статью, мнение экспертов в предметной области имеет значительно больший вес, чем мнение неспециалистов. SIQ™ статьи будет отображаться рядом со статьей после того, как она будет дважды оценена, и пересчитывается с каждой дополнительной оценкой.Посетите нашу страницу SIQ™, чтобы узнать больше.
Закрыть
Коэффициент научного влияния™ (SIQ™)
Scholarly Impact Quotient™ (SIQ™) — это наш уникальный процесс оценки рецензирования после публикации. SIQ™ оценивает важность и качество статей, используя коллективный разум сообщества Cureus в целом. Всем зарегистрированным пользователям предлагается внести свой вклад в SIQ™ любой опубликованной статьи. (Авторы не могут оценивать свои собственные статьи.)
У вас уже есть аккаунт? Войти.
Введите адрес электронной почты, чтобы получить бесплатную загрузку в формате PDF.
Обратите внимание, что тем самым вы соглашаетесь быть добавленным в наш ежемесячный список рассылки информационных бюллетеней по электронной почте.
Необычный случай глюкозо-галактозной мальабсорбции с Детская нейроаксональная дистрофия
¹Консультант по детской неврологии и эпилептологии, Кинг Специализированная больница Фахада, Саудовская Аравия
²Ассистент-консультант по детской неврологии, King Fahad Специализированная больница, Саудовская Аравия
* Автор, ответственный за переписку: Аль-Барадие Р.С., консультант Детский невролог и эпилептолог, педиатр Директор программы неврологии, специалист по королю Фахду Больница, Даммам, 31444, а/я 15215, Саудовская Аравия
Получено: 19 августа 2016 г.; Принято: 04 октября 2016 г .; Опубликовано: 07 октября 2016 г.
Реферат
Справочная информация: Глюкозо-галактозная мальабсорбция (ГГМ) является редкой аутосомно-рецессивное заболевание, проявляющееся диареей и задержкой развития в неонатальном периоде и при отсутствии лечения пациент может умереть из-за тяжелой обезвоживание и метаболический ацидоз.
Это заболевание возникает в результате мутации
ген, кодирующий котранспортер натрия/глюкозы на хромосоме 22. Инфантильный
Нейроаксональная дистрофия (INAD) — еще одно редкое аутосомно-рецессивное заболевание.
характеризуется аномальными нервными окончаниями либо в центральной нервной системе, либо в
периферическая нервная система. Пациенты обычно имеют нормальное развитие до 2
лет до начала прогрессирующей регрессии.Методы: Был проведен ретроспективный анализ карт.
Результаты: Молекулярно-генетический анализ был проведен для SLC5A1 и PLA2G6. гены. В обоих генах были обнаружены две разные гомозиготные мутации. подтверждающие диагнозы.
Заключение: Мы сообщаем о 8-летней девочке с диареей. в неонатальном периоде и в конечном итоге был диагностирован GGM. Позже у этого пациента была регрессия развития, а не признак GGM, для которого были проведены дополнительные исследования, которые подтвердили диагноз INAD.
К
Насколько нам известно, это первый в мире зарегистрированный случай наличия как
состояния у одного и того же пациента. Аналогичным образом пострадал ее двоюродный брат.Ключевые слова: Глюкозо-галактозная мальабсорбция; Инфантильный нейроаксональный дистрофия; Атрофия мозжечка
Введение
GGM — аутосомно-рецессивное заболевание с опасным для жизни диарея в период новорожденности, вызванная мутацией в системе Na+/глюкоза ген котранспортера SLC5A1. Пациенты с ГГМ имеют неонатальное начало тяжелой водянистой диареи, угрожающей жизни, и обезвоживание [1,2]. SLC5A1 является членом большого семейства генов, натрий: семейство растворенных симпортеров с геном SLC5A1, расположенным на хромосома 22q12.3, состоящая из 15 экзонов и кодирующая 73-кДа гликопротеину предсказывается наличие 14 трансмембранных сегментов [3]. После экспрессии белок SLC5A1 локализуется на щеточной кайме. оболочки кишечного эпителия и активно импортирует люминальные глюкоза или галактоза в энтероцит путем связывания транспорта сахара с градиентом Na+ через мембрану.
Мутации в SLC5A1 имеют
было показано, что они вызывают нарушение транспорта сахара [4].Подобно расширенной родословной амишей с большой когортой, водянистая диарея обычно начинается на 2-3 день после рождения, вскоре после начато грудное вскармливание или смесь. Диарея часто описывается родителями как «не останавливаясь», «продолжая бежать», «в каждом подгузник меняйте независимо от того, как часто вы меняете подгузник». Табурет кислая с pH около 5 или ниже, положительна на редуцирующие вещества. Другие лабораторные данные могут свидетельствовать о значительных метаболических нарушениях. ацидоз, гипернатриемическая и гиперосмолярная дегидратация [5]. Использовать безуглеводной смеси с фруктозой или без нее приводит к быстрая регидратация и нормальный рост и развитие после. Младенцы могут быть переведены с безуглеводной диеты на низкоуглеводную до 1 года, хотя у пациентов более частая дефекация (2-5 раз/сут) с жидким стулом по сравнению с нормальным. Толерантность к углеводсодержащим диета постепенно улучшается с течением времени у всех людей с разумным толерантность к регулярным углеводсодержащим диетам в подростковом возрасте.
Инфантильная нейроаксональная дистрофия (ИНАД) — заболевание, передающееся по аутосомно-рецессивному типу.
расстройство, вызванное мутациями в гене PLA2G6, расположенном на
хромосома 22q13.1, которая кодирует iPLA2-VIa, кальциевый
независимая фосфолипаза [6]. Ферменты iPLA2 имеют решающее значение
в гомеостазе клеточных мембран, так как они катализируют гидролиз
глицерофосфолипидов, образуя свободную жирную кислоту (обычно
арахидоновая кислота) и лизофосфолипид, которые могут лежать в основе
аксональная патология, наблюдаемая при заболевании, связанном с PLA2G6
[7]. Высокое содержание железа в мозге наблюдается примерно в половине случаев INAD, и это
является одним из многих заболеваний, известных как нейродегенерация головного мозга
накопления железа (НБЖА), группа прогрессирующих экстрапирамидных
нарушения, характеризующиеся накоплением железа в головном мозге.
появление симптомов обычно происходит до 2 лет, а психомоторное
регрессия является наиболее частым проявлением. Атаксия или неустойчивость походки
также часто встречается в начале заболевания. Атрофия зрительного нерва встречается у большинства
пораженных лиц, таких как нистагм и косоглазие [8]. Там
может быть ранняя туловищная гипотония с последующим развитием тетра
парез, обычно спастический, но может быть арефлексическим [9].]. Большинство
пациентов будут иметь признаки денервации на электромиограмме
(ЭМГ) и скорость нервной проводимости также могут быть снижены [9].
Сообщается о частом появлении быстрых ритмов на электроэнцефалограмме (ЭЭГ). В нескольких случаях сообщалось о генерализованных судорогах.
возникают на поздних стадиях заболевания. Железо в основном накапливается в глобусе.
pallidus, проявляющийся низкой интенсивностью сигнала на Т2-взвешенных изображениях. Рано
атрофия мозжечка и повышенная интенсивность сигнала в коре мозжечка
Взвешенные последовательности T2 являются обычным явлением. Диагноз подтверждается
мутация в ген. Сфероиды периферических нервов при биопсии нерва
сильно наводят на мысль о состоянии у пациентов с совместимыми
клинических условиях, если неинвазивный молекулярный анализ на мутацию
отрицательный. К сожалению, это состояние, ограничивающее жизнь, у большинства
больные умирают в первое десятилетие жизни из-за отсутствия специфического лечения
кроме симптоматического лечения баклофеном и ботулотоксином
для спастичности. Оба состояния наследуются по аутосомно-рецессивному типу.
и возможность каждого выше в сильно кровнородственном
населения. Поскольку специфического лечения в настоящее время не существует,
Симптоматическое лечение — это все, что предлагали междисциплинарные
команда. Задержка в диагностике приводит к задержке столь необходимого
генетическое консультирование, длительная неуверенность в диагнозе
лица, осуществляющие уход, и врач, а также ненужные исследования, даже если
состояние оказывается неизлечимым.Методы
Наш показательный случай родился от кровнородственных родителей первой степени родства из восточной части Саудовской Аравии (рис. 1) без происшествий. беременность в срок через экстренное кесарево сечение из-за дистресс плода с оценкой по шкале Апгар 8 и 10 баллов на 1 и 5 мин.
соответственно весом 2 кг. Вскоре ее госпитализировали в реанимацию.
после выписки с водянистой диареей в анамнезе и метаболическими
ацидоз, когда она была только на грудном молоке. У нее было положительное снижение
вещества в моче и стуле, и она была улучшена с помощью специальных
формула на основе фруктозы. Ей поставили диагноз ГГМ. Она была
выписан домой в удовлетворительном состоянии на лактозосодержащие др.
безуглеводная формула, и она чувствовала себя хорошо и чувствовала себя хорошо
после.Она начала переворачиваться к 6 месяцам и самостоятельно сидела на стуле. возраст 7 мес. В возрасте 7 месяцев у нее был фокальный припадок. в связи с чем ее госпитализировали и лечили в больнице. Мозг Сделана МРТ, которая показала инфаркт левой средней мозговой артерии с умеренная мозжечковая атрофия (рис. 2). Ее обширный метаболический и гиперкоагуляция была ничем не примечательна. Она продолжала иметь нормальные вехи развития до 2 лет, когда она начала терять способность сидеть, переворачиваться и контролировать голову.
К возрасту
3,5 года не могла сидеть, ползать, говорить, были трудности в
зрение. В конце болезни она потеряла слух. После этого она начала
иметь трудности с кормлением при тяжелой ГЭРБ и гастростоме
была установлена в возрасте 4 лет вместе с фундопликацией. Один
год спустя у нее диагностировали нейроаксональную дистрофию. мышцы
биопсия показала денервационную атрофию. У нее было несколько госпитализаций
больнице из-за аспирационной пневмонии, и она следует с
мультидисциплинарная команда. При медицинском осмотре ее вес был 18.
кг и ее рост 110 см в 8 лет. У нее был гипотонус и плохое
контроль головы и туловища с множественными деформациями обеих стоп. Она также
были контрактуры запястий. У нее был нистагм, грудопоясничный
сколиоз вправо.Последний раз ее госпитализировали в конце 2015 года, когда ей было 8 лет. Она была госпитализирована в отделение интенсивной терапии с тяжелой пневмонией в анамнезе. скончался в результате септического шока. У нее есть двоюродный брат, которому сейчас 8,5 лет, у которого похожее заболевание.
У него эпилепсия под контролем
только вальпроат в течение последних 2 лет, не говорит и не передвигается.Результаты
Ген SLC5A1 анализировали с помощью ПЦР (полимеразная цепная Реакция) и секвенирование обеих цепей ДНК всей кодирующей область и высококонсервативные экзон-интронные соединения сплайсинга. Она имел гомозиготный c.265G>A (стр. G89R) вариант в экзоне 3 Ген SLC5A1 на хромосоме 22q12.3. Эта конкретная мутация в ген SLC5A1 ранее в литературе не описывался. это в высококонсервативном положении нуклеотида и аминокислоты. Программного обеспечения Анализ Polyphen, SIFT и AGVGD показывает, что изменение вероятно вредные (CGC Genetics, Лиссабон, Португалия). Ген PLA2G6 анализ состоял из ПЦР-амплификации и прямого секвенирования вся кодирующая область гена со всеми соответствующими интрон-экзонными границы. Гомозиготный c.2070_2072del (стр. Val691дел) мутация, ранее описанный в литературе, был обнаружен в PLA2G6 на хромосома 22q13.1, которая не изменяет рамку считывания ген, подтверждающий диагноз INAD.
Обсуждение
Мы сообщаем об индексном случае GGM с INAD, с подобными состояние ее двоюродного брата. Мутация в гене SLC5A1 была новый и пациент ответил на специальную формулу на основе фруктозы нормально развивается до 2 лет. Несколько различных мутаций в SLC5A1, вызывающем GGM, включают промах, бессмыслицу, сдвиг кадра, сайт сплайсинга и промоторные мутации. Бессмыслица, сдвиг кадра и Все мутации сайта сплайсинга продуцируют нефункциональные укороченные белки. В большинстве миссенс-мутаций белки транслируются и стабильны в клетке, но не достигают плазматической мембраны [10]. Кишечный котранспортер натрия/глюкозы отвечает за «активное» поглощение глюкозы через мембрану щеточной каемки клетки, выстилающие желудочно-кишечный тракт. это энергозатратный действие, которое управляется натрий/калиевой АТФазой, расположенной в базолатеральной клеточной мембраны. Трансэпителиальное всасывание глюкоза и галактоза затем завершаются на базально-латеральной мембране. через облегченный переносчик глюкозы.
Интригующие симптомы психомоторной регрессии, которые не является частью GGM, что привело к поиску другого сосуществующего условия. После с опозданием на 3 года у нее было подтверждено наличие INAD на основании ее описанная выше мутация в гене PLA2G6. Этот конкретный мутация описана в литературе [11]. Самое интересное следует отметить непосредственную близость этих двух генов в области 22q12.3 и 22q13.1 соответственно, хотя обе являются точечными мутациями, а не случаем последовательной делеции генов.
Состояние гиперкоагуляции и/или инсульты ранее не описаны как признаки одного из двух состояний. Наличие левого среднего инфаркт мозговой артерии обнаружен в возрасте 7 месяцев после того, как она поступила с фокальными припадками привели к детальной оценке. Она соответствовала диета и не было эпизодов диареи или обезвоживания в то время начала приступа, и был тщательно исследован на предмет любого известного состояние гиперкоагуляции без каких-либо сведений о причине ее инсульта.
это
трудно с какой-либо степенью точности комментировать время
инсульт, так как первая МРТ головного мозга показала старый инфаркт, и, следовательно, она была
маркируется как внутриутробный инсульт, хотя возможно обезвоживание в отдаленном прошлом с инфарктом.Приступы у пациентов с НАД поздние и легко контролируемые, но у нашего пациента был ранний приступ, вероятно, в результате инсульта. Ей У двоюродного брата обычно были поздние приступы приступов в возрасте 6 лет, и у обоих выраженная мозжечковая атрофия, описанная в предыдущих отчетах о случаях.
Ссылки
- Райт Э.М., Мартин М.Г., Терк. Семейная глюкозо-галактозная мальабсорбция и наследственная почечная глюкозурия. В: ScriverCR, BeaudeTAL, SlyWS, ValleD, ред. Метаболические и молекулярные основы наследственных заболеваний, Vol. 3. Нью-Йорк: Макгроу-Хилл, 2001: 489.1-4908.
- Райт Э.М. I. Мальабсорбция глюкозы-галактозы. Am J Physiol. 1998 год; 275: G879-G882.
- Терк Э., Райт Э.М. Мотивы мембранной топологии в котранспортере SGLT
семья. J Membr Biol. 1997 год; 159: 1-20.
- Райт Э.М., Терк Э., Мартин М.Г. Молекулярная основа глюкозо-галактозы нарушение всасывания. Клеточная биохимия Биофиз. 2002 г.; 36: 115-121.
- Xin B, Wang H. Множественные вариации последовательности в гене SLC5A1 связаны с глюкозо-галактозной мальабсорбцией в большой когорте амишей старого порядка. Клин Жене. 2011 г.; 79: 86-91.
- Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S. PLA2G6, кодирующий фосфолипазу А2, мутирует при нейродегенеративном заболевании. расстройства с высоким содержанием железа в головном мозге. Нат Жене. 2006 г.; 38: 752-754.
- Бабурина И., Яковски С. Клеточные реакции на избыток фосфолипидов. Джей Биол хим. 1999 г.; 274: 9400-9408.
- Куриан М.А., Морган Н.В., Макферсон Л., Фостер К., Пик Д., Гупта Р. Фенотипический спектр нейродегенерации, связанной с мутациями в Ген PLA2G6 (PLAN). Неврология. 2008 г.; 70:1623-1629.
- Нардоччи Н., Зорзи Г., Фарина Л.
- Rafeey M, Golzar A: Синдром глюкозо-галактозной мальабсорбции, проявляющийся врожденной диареей.
 Питательные вещества. 2016, 8:157. 10.3390/nu8030157
Питательные вещества. 2016, 8:157. 10.3390/nu8030157 Пак J Med Sci. 2007, 23:959-61.
Пак J Med Sci. 2007, 23:959-61. Альфараиди
Альфараиди Конфликт интересов: В соответствии с единой формой раскрытия информации ICMJE все авторы заявляют следующее: Информация об оплате/услугах: Все авторы заявили, что никакая финансовая поддержка представленной работы не была получена ни от одной организации. Финансовые отношения: Все авторы заявили, что у них нет финансовых отношений в настоящее время или в течение предыдущих трех лет с какими-либо организациями, которые могут быть заинтересованы в представленной работе. Другие отношения: Все авторы заявили об отсутствии других отношений или действий, которые могли бы повлиять на представленную работу.
Конфликт интересов: В соответствии с единой формой раскрытия информации ICMJE все авторы заявляют следующее: Информация об оплате/услугах: Все авторы заявили, что никакая финансовая поддержка представленной работы не была получена ни от одной организации. Финансовые отношения: Все авторы заявили, что у них нет финансовых отношений в настоящее время или в течение предыдущих трех лет с какими-либо организациями, которые могут быть заинтересованы в представленной работе. Другие отношения: Все авторы заявили об отсутствии других отношений или действий, которые могли бы повлиять на представленную работу. С. и др. (02 октября 2021 г.) Врожденная глюкозо-галактозная мальабсорбция: случай с новой мутацией SLC5A1 у младенца из Саудовской Аравии. Куреус 13(10): e18440. Дои: 10.7759/cureus.18440
С. и др. (02 октября 2021 г.) Врожденная глюкозо-галактозная мальабсорбция: случай с новой мутацией SLC5A1 у младенца из Саудовской Аравии. Куреус 13(10): e18440. Дои: 10.7759/cureus.18440
 Все, что выше 5, следует считать выше среднего. Хотя все зарегистрированные пользователи Cureus могут оценивать любую опубликованную статью, мнение экспертов в предметной области имеет значительно больший вес, чем мнение неспециалистов. SIQ™ статьи будет отображаться рядом со статьей после того, как она будет дважды оценена, и пересчитывается с каждой дополнительной оценкой.
Все, что выше 5, следует считать выше среднего. Хотя все зарегистрированные пользователи Cureus могут оценивать любую опубликованную статью, мнение экспертов в предметной области имеет значительно больший вес, чем мнение неспециалистов. SIQ™ статьи будет отображаться рядом со статьей после того, как она будет дважды оценена, и пересчитывается с каждой дополнительной оценкой.
 Это заболевание возникает в результате мутации
ген, кодирующий котранспортер натрия/глюкозы на хромосоме 22. Инфантильный
Нейроаксональная дистрофия (INAD) — еще одно редкое аутосомно-рецессивное заболевание.
характеризуется аномальными нервными окончаниями либо в центральной нервной системе, либо в
периферическая нервная система. Пациенты обычно имеют нормальное развитие до 2
лет до начала прогрессирующей регрессии.
Это заболевание возникает в результате мутации
ген, кодирующий котранспортер натрия/глюкозы на хромосоме 22. Инфантильный
Нейроаксональная дистрофия (INAD) — еще одно редкое аутосомно-рецессивное заболевание.
характеризуется аномальными нервными окончаниями либо в центральной нервной системе, либо в
периферическая нервная система. Пациенты обычно имеют нормальное развитие до 2
лет до начала прогрессирующей регрессии. К
Насколько нам известно, это первый в мире зарегистрированный случай наличия как
состояния у одного и того же пациента. Аналогичным образом пострадал ее двоюродный брат.
К
Насколько нам известно, это первый в мире зарегистрированный случай наличия как
состояния у одного и того же пациента. Аналогичным образом пострадал ее двоюродный брат. Мутации в SLC5A1 имеют
было показано, что они вызывают нарушение транспорта сахара [4].
Мутации в SLC5A1 имеют
было показано, что они вызывают нарушение транспорта сахара [4]. Инфантильная нейроаксональная дистрофия (ИНАД) — заболевание, передающееся по аутосомно-рецессивному типу.
расстройство, вызванное мутациями в гене PLA2G6, расположенном на
хромосома 22q13.1, которая кодирует iPLA2-VIa, кальциевый
независимая фосфолипаза [6]. Ферменты iPLA2 имеют решающее значение
в гомеостазе клеточных мембран, так как они катализируют гидролиз
глицерофосфолипидов, образуя свободную жирную кислоту (обычно
арахидоновая кислота) и лизофосфолипид, которые могут лежать в основе
аксональная патология, наблюдаемая при заболевании, связанном с PLA2G6
[7]. Высокое содержание железа в мозге наблюдается примерно в половине случаев INAD, и это
является одним из многих заболеваний, известных как нейродегенерация головного мозга
накопления железа (НБЖА), группа прогрессирующих экстрапирамидных
нарушения, характеризующиеся накоплением железа в головном мозге.
появление симптомов обычно происходит до 2 лет, а психомоторное
регрессия является наиболее частым проявлением. Атаксия или неустойчивость походки
также часто встречается в начале заболевания.
Инфантильная нейроаксональная дистрофия (ИНАД) — заболевание, передающееся по аутосомно-рецессивному типу.
расстройство, вызванное мутациями в гене PLA2G6, расположенном на
хромосома 22q13.1, которая кодирует iPLA2-VIa, кальциевый
независимая фосфолипаза [6]. Ферменты iPLA2 имеют решающее значение
в гомеостазе клеточных мембран, так как они катализируют гидролиз
глицерофосфолипидов, образуя свободную жирную кислоту (обычно
арахидоновая кислота) и лизофосфолипид, которые могут лежать в основе
аксональная патология, наблюдаемая при заболевании, связанном с PLA2G6
[7]. Высокое содержание железа в мозге наблюдается примерно в половине случаев INAD, и это
является одним из многих заболеваний, известных как нейродегенерация головного мозга
накопления железа (НБЖА), группа прогрессирующих экстрапирамидных
нарушения, характеризующиеся накоплением железа в головном мозге.
появление симптомов обычно происходит до 2 лет, а психомоторное
регрессия является наиболее частым проявлением. Атаксия или неустойчивость походки
также часто встречается в начале заболевания. Атрофия зрительного нерва встречается у большинства
пораженных лиц, таких как нистагм и косоглазие [8]. Там
может быть ранняя туловищная гипотония с последующим развитием тетра
парез, обычно спастический, но может быть арефлексическим [9].]. Большинство
пациентов будут иметь признаки денервации на электромиограмме
(ЭМГ) и скорость нервной проводимости также могут быть снижены [9].
Сообщается о частом появлении быстрых ритмов на электроэнцефалограмме (ЭЭГ). В нескольких случаях сообщалось о генерализованных судорогах.
возникают на поздних стадиях заболевания. Железо в основном накапливается в глобусе.
pallidus, проявляющийся низкой интенсивностью сигнала на Т2-взвешенных изображениях. Рано
атрофия мозжечка и повышенная интенсивность сигнала в коре мозжечка
Взвешенные последовательности T2 являются обычным явлением. Диагноз подтверждается
мутация в ген. Сфероиды периферических нервов при биопсии нерва
сильно наводят на мысль о состоянии у пациентов с совместимыми
клинических условиях, если неинвазивный молекулярный анализ на мутацию
отрицательный.
Атрофия зрительного нерва встречается у большинства
пораженных лиц, таких как нистагм и косоглазие [8]. Там
может быть ранняя туловищная гипотония с последующим развитием тетра
парез, обычно спастический, но может быть арефлексическим [9].]. Большинство
пациентов будут иметь признаки денервации на электромиограмме
(ЭМГ) и скорость нервной проводимости также могут быть снижены [9].
Сообщается о частом появлении быстрых ритмов на электроэнцефалограмме (ЭЭГ). В нескольких случаях сообщалось о генерализованных судорогах.
возникают на поздних стадиях заболевания. Железо в основном накапливается в глобусе.
pallidus, проявляющийся низкой интенсивностью сигнала на Т2-взвешенных изображениях. Рано
атрофия мозжечка и повышенная интенсивность сигнала в коре мозжечка
Взвешенные последовательности T2 являются обычным явлением. Диагноз подтверждается
мутация в ген. Сфероиды периферических нервов при биопсии нерва
сильно наводят на мысль о состоянии у пациентов с совместимыми
клинических условиях, если неинвазивный молекулярный анализ на мутацию
отрицательный. К сожалению, это состояние, ограничивающее жизнь, у большинства
больные умирают в первое десятилетие жизни из-за отсутствия специфического лечения
кроме симптоматического лечения баклофеном и ботулотоксином
для спастичности. Оба состояния наследуются по аутосомно-рецессивному типу.
и возможность каждого выше в сильно кровнородственном
населения. Поскольку специфического лечения в настоящее время не существует,
Симптоматическое лечение — это все, что предлагали междисциплинарные
команда. Задержка в диагностике приводит к задержке столь необходимого
генетическое консультирование, длительная неуверенность в диагнозе
лица, осуществляющие уход, и врач, а также ненужные исследования, даже если
состояние оказывается неизлечимым.
К сожалению, это состояние, ограничивающее жизнь, у большинства
больные умирают в первое десятилетие жизни из-за отсутствия специфического лечения
кроме симптоматического лечения баклофеном и ботулотоксином
для спастичности. Оба состояния наследуются по аутосомно-рецессивному типу.
и возможность каждого выше в сильно кровнородственном
населения. Поскольку специфического лечения в настоящее время не существует,
Симптоматическое лечение — это все, что предлагали междисциплинарные
команда. Задержка в диагностике приводит к задержке столь необходимого
генетическое консультирование, длительная неуверенность в диагнозе
лица, осуществляющие уход, и врач, а также ненужные исследования, даже если
состояние оказывается неизлечимым. соответственно весом 2 кг. Вскоре ее госпитализировали в реанимацию.
после выписки с водянистой диареей в анамнезе и метаболическими
ацидоз, когда она была только на грудном молоке. У нее было положительное снижение
вещества в моче и стуле, и она была улучшена с помощью специальных
формула на основе фруктозы. Ей поставили диагноз ГГМ. Она была
выписан домой в удовлетворительном состоянии на лактозосодержащие др.
безуглеводная формула, и она чувствовала себя хорошо и чувствовала себя хорошо
после.
соответственно весом 2 кг. Вскоре ее госпитализировали в реанимацию.
после выписки с водянистой диареей в анамнезе и метаболическими
ацидоз, когда она была только на грудном молоке. У нее было положительное снижение
вещества в моче и стуле, и она была улучшена с помощью специальных
формула на основе фруктозы. Ей поставили диагноз ГГМ. Она была
выписан домой в удовлетворительном состоянии на лактозосодержащие др.
безуглеводная формула, и она чувствовала себя хорошо и чувствовала себя хорошо
после. К возрасту
3,5 года не могла сидеть, ползать, говорить, были трудности в
зрение. В конце болезни она потеряла слух. После этого она начала
иметь трудности с кормлением при тяжелой ГЭРБ и гастростоме
была установлена в возрасте 4 лет вместе с фундопликацией. Один
год спустя у нее диагностировали нейроаксональную дистрофию. мышцы
биопсия показала денервационную атрофию. У нее было несколько госпитализаций
больнице из-за аспирационной пневмонии, и она следует с
мультидисциплинарная команда. При медицинском осмотре ее вес был 18.
кг и ее рост 110 см в 8 лет. У нее был гипотонус и плохое
контроль головы и туловища с множественными деформациями обеих стоп. Она также
были контрактуры запястий. У нее был нистагм, грудопоясничный
сколиоз вправо.
К возрасту
3,5 года не могла сидеть, ползать, говорить, были трудности в
зрение. В конце болезни она потеряла слух. После этого она начала
иметь трудности с кормлением при тяжелой ГЭРБ и гастростоме
была установлена в возрасте 4 лет вместе с фундопликацией. Один
год спустя у нее диагностировали нейроаксональную дистрофию. мышцы
биопсия показала денервационную атрофию. У нее было несколько госпитализаций
больнице из-за аспирационной пневмонии, и она следует с
мультидисциплинарная команда. При медицинском осмотре ее вес был 18.
кг и ее рост 110 см в 8 лет. У нее был гипотонус и плохое
контроль головы и туловища с множественными деформациями обеих стоп. Она также
были контрактуры запястий. У нее был нистагм, грудопоясничный
сколиоз вправо.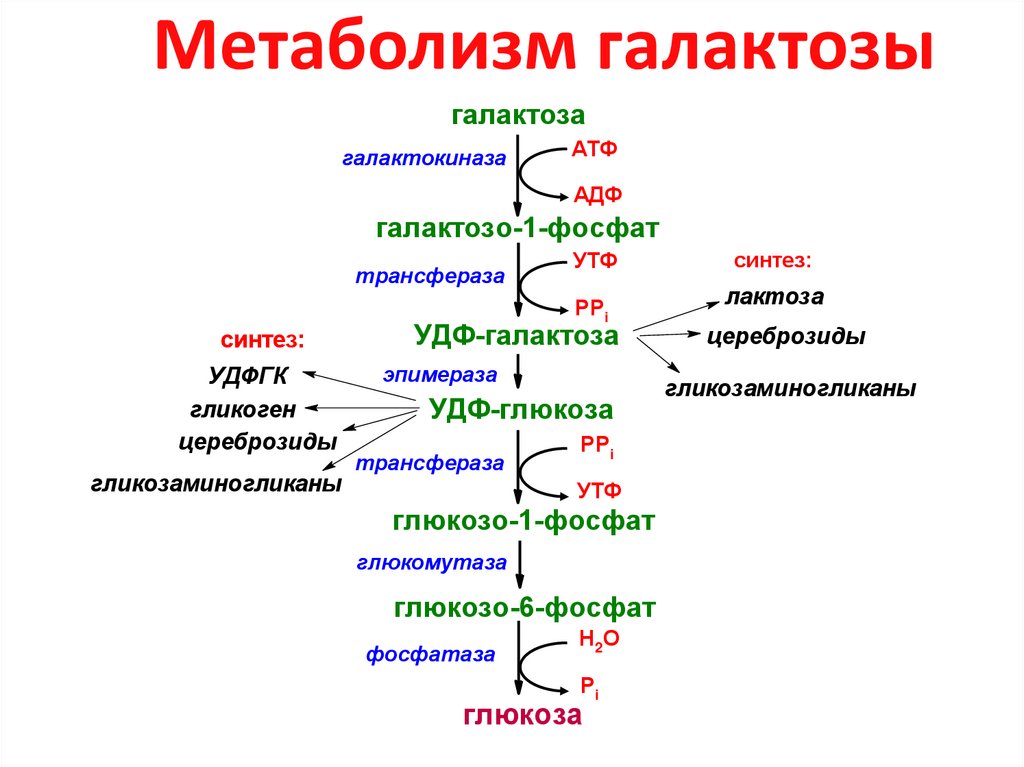 У него эпилепсия под контролем
только вальпроат в течение последних 2 лет, не говорит и не передвигается.
У него эпилепсия под контролем
только вальпроат в течение последних 2 лет, не говорит и не передвигается.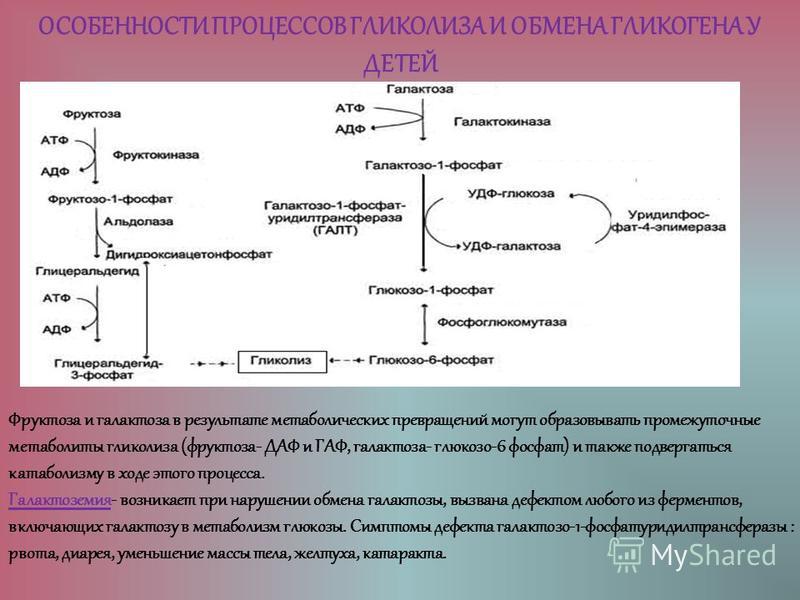

 это
трудно с какой-либо степенью точности комментировать время
инсульт, так как первая МРТ головного мозга показала старый инфаркт, и, следовательно, она была
маркируется как внутриутробный инсульт, хотя возможно обезвоживание в отдаленном прошлом с инфарктом.
это
трудно с какой-либо степенью точности комментировать время
инсульт, так как первая МРТ головного мозга показала старый инфаркт, и, следовательно, она была
маркируется как внутриутробный инсульт, хотя возможно обезвоживание в отдаленном прошлом с инфарктом. J Membr Biol. 1997 год; 159: 1-20.
J Membr Biol. 1997 год; 159: 1-20.